PEG-BIO Biopharm Co.,Ltd(chongqing).


News Center
31 monoclonal antibodies to watch for in 2018
Source:药事纵横
|
Author:pro954f45
|
Published time: 2018-01-14
|
2979 Views
|
Share:
overview
Monoclonal Antibody (Mab). Since the first Monoclonal antibody was introduced in 1986, nearly 80 Monoclonal antibodies have been published worldwide. So far, MONOclonal antibody has been developed to the fourth generation. The first generation is momAB, the second generation is ximab, the third generation is Zumab, and the fourth generation is MUMab. The advantage of humanized monoclonal antibody is that it can overcome the reaction of human anti-mouse antibody, avoid the rapid elimination of monoclonal antibody molecules as foreign proteins by the immune system, and improve the biological activity of monoclonal antibody molecules.

Monoclonal Antibody (Mab). Since the first Monoclonal antibody was introduced in 1986, nearly 80 Monoclonal antibodies have been published worldwide. So far, MONOclonal antibody has been developed to the fourth generation. The first generation is momAB, the second generation is ximab, the third generation is Zumab, and the fourth generation is MUMab. The advantage of humanized monoclonal antibody is that it can overcome the reaction of human anti-mouse antibody, avoid the rapid elimination of monoclonal antibody molecules as foreign proteins by the immune system, and improve the biological activity of monoclonal antibody molecules.
(Image from the Internet)
Despite the small number of products, there are more than 40 active monoclonal antibodies that support a global market of nearly $100 billion, and the growth rate of the drug market for monoclonal antibodies is far higher than that of other drugs in the industry. Excellent curative effect, huge market and high added value have given rise to a large number of companies developing monoclonal antibody products. In recent years, the development speed of the global monoclonal antibody industry has been significantly accelerated. In 2017, the first monoclonal antibodies approved in the US and Europe reached double-digit levels for the first time, and 2018 is expected to see further highs. As of December 1, 2017, nine monoclonal antibodies are under FDA/EMA review, and 12 have received or will receive critical clinical data, and are expected to file for marketing by 2018. In addition, there are nearly 20 monoclonal antibody products at the terminal clinical stage. In the near future, it will be an era of "hundred ships".
Review of approved MONOclonal antibodies in Europe and the US in 2017
As of December 1, 2017, 10 monoclonal antibodies were approved for the first time in the United States and Europe. With the exception of Brodalumab, which was approved by Japan in 2016, and Sarilumab, which was approved by Canada in January 2017, all products are among the first in the world. These are 10 monoclonal antibodies, five are immune, four are anti-tumor, and one is blood disease.

Review of approved MONOclonal antibodies in Europe and the US in 2017
As of December 1, 2017, 10 monoclonal antibodies were approved for the first time in the United States and Europe. With the exception of Brodalumab, which was approved by Japan in 2016, and Sarilumab, which was approved by Canada in January 2017, all products are among the first in the world. These are 10 monoclonal antibodies, five are immune, four are anti-tumor, and one is blood disease.
Brodalumab
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827) is a human IgG2 antibody, targeting the interleukin-17 receptor (IL-17RA). It can block the inflammatory signal of IL-17A through IL-17RA, and promote the inflammatory cytokines IL-17F and IL-17C. Brodalumab was first approved in Japan on July 4, 2016 for Lumicef, which includes erythrodermic psoriasis, pustular psoriasis, psoriatic arthritis and psoriasis vulgaris. In February 2017 and July 2017, Brodalumab was approved in the United States and Europe, respectively, for the treatment of moderate to severe plaque psoriasis in adults who do not respond to systemic therapy or light therapy (UV therapy). Safety and efficacy of Brodalumab was studied in three placebo-controlled phase III clinical trials, amagine-1 (NCT01708590), Amagine-2 (NCT01708603) and Amagine-3 (NCT01708603), involving a total of 4,373 patients. The primary end point was the proportion of patients with psoriasis at week 12 who improved by more than 75% in area with severity (PASI75) and whose physician's static overall assessment (sPGA) score (0/1) decreased by at least 2 points from baseline. Amagine-1 results showed that 83 per cent of Brodalumab 210mg to PASI75, 60 per cent of the 140mg group and 3 per cent of the placebo group. The sPGA scores were also higher than the placebo group at 76%, 54% and 1%, respectively. Amagine-2 and Amagine-3 test data showed that the improvement of clinical symptoms of moderate and severe psoriasis was significantly better than that of placebo group. In amagine-2, the PASI compliance rate was 86% in 210mg group, 67% in 140mg group, and 8% in placebo group, 86% in Amagine-3, 69% and 6% in amagine-3, respectively. The sPGA compliance rate was also higher in the treatment group, 79% in 210mg amagine-2, 58% in 140mg group, and 4% in placebo group. In Amagine-3, 80%, 60% and 4%, respectively.
Avelumab
Avelumab (Bavencio, MSB0010718C), a human IgG1 monoclonal antibody targeting PD-L1, was first approved on May 23, 2017. The indications approved were metastatic Merkel-cell carcinoma in adults and children over 12 years of age. The indication was also approved in Europe in September 2017. Avelumab was approved based on data from the Javelin Merkel 200 study (NCT02155647), A key phase II clinical trial in which Part A patients underwent chemotherapy prior to initial treatment with Avelumab in Part B. At a dose of 10mg/kg, intravenous injection is given fortnightly. In Part A, 88 patients who had undergone prior chemotherapy were treated with this product and their conditions were controlled. The median follow-up period was 10.4 months, and the objective remission rate was 31.8%, among which 8% had complete remission and 20% had partial remission. In Part B, 39 patients were treated for the first time with this product, and data on the median remission time of the primary endpoint is expected to be available in September 2019. Avelumab has previously been granted orphan status for Merckle cell cancer in Europe and Australia, as well as FDA breakthrough therapy designation. On May 9, 2017, THE FDA granted Avelumab a priority review of indications for locally advanced or metastatic urothelial carcinoma that has progressed after platinum chemotherapy. In addition, four phase III clinical trials of the product for non-small cell lung cancer, renal cell cancer, ovarian cancer and gastric cancer will be completed in 2018 and have been awarded orphan drug status for gastric cancer in both the United States and Europe.
Dupilumab
Dupilumab (Dupixent, REGN668/SAR231893), an IL4R-targeted IgG4 monoclonal antibody, was approved by the FDA on April 28, 2017, and by THE EMA on September 28, 2017, respectively, for the treatment of atopic dermatitis. Dupixent's approval was based on LIBERTY, an atopic dermatitis study that included three clinical trials of SOLO1, SOLO2 and CHRONOS. In SOLO1 and SOLO2, patients were treated with either 300mg Dupixent or placebo once a week, or with 300mg Dupixent every two weeks, respectively, and switched to placebo. The efficacy of Dupixent combined with corticosteroid was studied by CHRONOS test with placebo as control. The primary end point of the trial was the proportion of patients with a reduction of more than two points from baseline in the investigator's Overall Evaluation (IGA) score (0/1) at week 16. The results showed that, after 16 weeks of Dupixent monotherapy, the IGA compliance was significantly higher than that of the placebo group, with 37% of SOLO1 and 36% of SOLO2. Similarly, by CHRONOS, the dupilumab+ corticosteroid combination treatment was 39% and the placebo + steroid combination treatment was 12%. Because of its outstanding efficacy, this product has been recognized by FDA as a breakthrough therapy for atopic dermatitis. In addition to atopic dermatitis, Dupilumab is currently in phase III clinical development for indications for asthma and nasal polyps. In September 2017, Sanofi/Regeneration announced that dupilumab's LIBERTY ASTHMA QUEST (NCT02414854) trial for uncontrolled persistent ASTHMA had reached its therapeutic endpoint. Two other clinical trials on nasal polyp treatment (NCT02912468, NCT02898454) will also reach their endpoints in 2018. More encouragingly, this product can also be used in the treatment of eosinophilic esophagitis, and the current research is in the clinical phase II, and has been recognized as an orphan drug by FDA.
Ocrelizumab
Ocrelizumab (Ocrevus) is a humanized IgG1 monoclonal antibody that targets Cd-20-positive B cells. These B cells play a role in myelin damage and multiple sclerosis by producing pro-inflammatory cytokines, secreting autoantibodies, and activating pro-inflammatory T cells. Ocrevus destroys CD20 positive B cells in three main ways: complement dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity, and by binding to CD20 on target cells to apoptosis. This monoclonal antibody has been studied in both internal rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), but the expected results are not satisfactory. Ocrevus received FDA approval on March 29 for relapsing and relapsing multiple sclerosis as well as primary progressive multiple sclerosis, following the indication's FDA breakthrough therapy designation, fast-track and priority review. Two trials, OPERAI (NCT01247324) and OPERAII (NCT01412333), respectively, assessed the safety and efficacy of Ocrevus in the treatment of multiple sclerosis with remission, and showed significantly lower recurrence rates in the Ocrevus group than in the beta interferon group, with OPERAI 46% lower and OPERAII 57% lower. In primary progressive multiple sclerosis, results from the ORATORIO trial showed that Ocrevus significantly reduced clinical disability progression at 12 weeks (primary endpoint) compared with placebo (32.9% vs 39.3%). In addition, MRI showed a 3.4 percent decrease in the overall volume of encephalopathy in patients treated with Ocrevus, compared with a 7.4 percent increase in placebo.
Durvalumab
Durvalumab (Imfinzi, MEDI4736) is a human IgG1 antibody that directly activates PD-L1 to block the association between PD-L1 and its receptor (PD-1, CD80). Durvalumab, designed to block the interaction between cytotoxic effects and PD-L1 positive immune cells, was approved by the FDA on May 1, 2017 for accelerated use in locally advanced or metastatic uEPITHELIAL carcinoma that is exacerbated by platinum chemotherapy. This is a combination of priority review and breakthrough therapy, based on a phase 1/2 clinical trial of 182 patients (NCT01693562). Durvalumab was given intravenously every two weeks at a dose of 10mg/kg for 12 months, and the objective remission rate was 27.4% in the subgroup with high EXPRESSION of PD-L1 and 4.1% in the subgroup with low expression of PD-L1. In addition, the NDA for locally advanced or inoperable non-small cell lung cancer (NSCLC) after platinum chemotherapy has entered the FDA review stage, having previously obtained FDA priority review and breakthrough therapy recognition. At the same time, an application for marketing permission (MAA) for this indication has been accepted by EMA, and data from the phase III PACIFI clinical trial (NCT02125461) has also been submitted to EMA. Clinical results showed that for NSCLC progressing after chemotherapy, the median progression-free survival was 16.8 months, while the placebo was only 5.6 months. In addition to the above two indications, Durvalumab has been granted fast-track status by FDA for the treatment of pD-L1-positive metastatic head and neck cancer. Phase III clinical trials EAGLE (NCT02369874) and KESTREL (NCT02551159) are expected to reach their endpoints in February and March 2018, respectively.
Sarilumab
Sarilumab (Kevzara SAR153191, REGN88) is a kind of IL - 6 r as targets of IgG1 sheet resistance, in February 2017, approved, for the first time in Canada in May 2017 and July 2017 respectively and cleared by the FDA and EMA for one or more biological products or biological products regulatory anti-rheumatism disease drugs (DMARDs) inadequate response or intolerance of the relation between active rheumatoid arthritis. Sarilumab's approval was based on a global multicenter Sari-RA clinical trial involving MOBILITY (NCT01061736), TARGET (NCT01709578) and MONARCH (NCT02332590). Sarilumab was used in combination with methotrexate or other DMARD in both the MOBILITY and TARGET trials. Results of the MOBILITY study showed that ACR20 at 24 weeks was 66% in the 200mg sarilumab treatment group, 58% in the 150mg group, and only 33% in the placebo group. Sarilumab also improved patients' physiological function. Haq-di score decreased by 0.58 in the 200mg Sarilumab treatment group, 0.54 in the 150mg group, and 0.30 in the placebo group. TARGET showed similar results: 61% of ACR20 in the DMARD+200mg Sarilumab group, 56% in the 150mg group, and 34% in the placebo group. In terms of physiological function improvement, haq-DI score decreased by 0.50 in the DMARD+200mg Sarilumab treatment group and 0.49 in the 150mg group, while only 0.29 in the placebo group. The MONARCH test assessed the safety and effectiveness of Sarilumab against adamumab. At 24 weeks, the disease activity score (DAS28-ESR) for 28 joints calculated using ERYTHROcyte sedimentation rate decreased by 3.28 from baseline, while the adamumab group decreased by only 2.20, which was also the primary endpoint.
Guselkumab
Guselkumab (Tremfya) is a human IgG1, which blocks the P19 subunit of IL-23, thereby limiting inflammatory response. Il-23 is a cytokine involved in Th17 cell differentiation and maintenance, producing pro-inflammatory cytokines IL-17, IL-22 and TNF through T cell subsets. On July 13, 2017, Guselkumab received FDA approval for moderate to severe plaque psoriasis for systemic therapy (injection or oral treatment) or phototherapy (ultraviolet treatment), and in November 2017, guselkumab was reapproved by EMA for the same indications as in the United States. Guselkumab was granted based on the results of three clinical trials, VOYAGE1 (NCT02207231), VOYAGE2 (NCT02207244) and NAVIGATE (NCT02203032). VOYAGE1 and VOYAGE2 evaluated the efficacy and safety of Tremfya relative to placebo and altamumab. The results showed that after 16 weeks of guselkumab treatment, the IGA compliance rate was far higher than that of the placebo group (85.1% vs 6.9%). In addition, 73.3% and 70.0% of the patients in the Tremfya treatment group achieved PASI90 remission, while 49.7% and 46.8% of the altamumab treatment group achieved relief, respectively. The NAVIGATE test evaluates the efficacy and safety of Tremfya versus Stelara. Patients receive Ustekinumab at week 0 and 4, and patients with inadequate response are given ustekinumab or guselkumab again at week 16. The primary end point is the proportion of patients whose IGA score has decreased by a minimum of 2 points at week 28 from baseline. The results showed that after treatment with ustekinumab or guselkumab, the IGA score reaching rates were 14.3% and 31.1% at 28 weeks, and 17.3% and 36.3% at 32 weeks, respectively. The data indicate that Tremfya still has significant therapeutic effect in patients with inadequate response to previous treatment with Stelara.
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa, CMC-544) is a conjugate of humanized IgG4 antibody against CD22 and the cytotoxic drug Kazamycin. Besponsa was first approved in Europe on June 30, 2017 and again approved by THE FDA on August 17, 2017 for both adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL). Prior to this, Besponsa has been approved as an orphan drug in Europe and the United States, as well as FDA's ALL Breakthrough Therapy designation. Besponsa was approved on the basis of a phase III clinical trial called INOVATE ALL (NCT01564784), which is controlled with existing standard therapies. Patients received three doses of Besponsa or standard chemotherapy on day 1 (0.8mg/m2), day 8 (0.5mg/m2), and day 15 (0.5mg/m2). The initial course is 21 days, and then each course is 28 days, leading to 6 courses. The results showed that the Besponsa group had a complete response rate of 80.7% and a median total survival time of 7.7 months, while the standard chemotherapy group had a complete response rate of 29.4% and a median total survival time of 6.7 months.
Benralizumab
Benralizumab (Fasenra, Medi-563) is a defucified IgG1 monoclonal antibody targeting the subunit of IL-5R. On November 14, 2017, THE FDA approved this product for additional maintenance therapy in patients with severe asthma aged 12 years and older with eosinophilic phenotypes, and on November 10, 2017, EMA's expert committee also gave a positive opinion on the release of this product. The safety and effectiveness of this product was evaluated in the WINDWARD study, which included 6 phase III clinical trials, including SIROCCO, CALIMA, ZONDA, BISE, BORA and GREGALE. Two randomized, double-blind, parallel, placebo-controlled clinical trials, SIROCCO(NCT01928771) and CALIMA (NCT01914757), evaluated the long-term safety of benralizumab. Patients were given a fixed subcutaneous injection of 30mg for 56 weeks, and the results of the study were published in 2016. In the 28-week ZONDA(NCT02075255) trial, patients with moderate to severe asthma were treated with either Benralizumab 30mg or placebo once every four or eight weeks on oral corticosteroids. The results showed a 75% reduction in corticosteroids in the Benralizumab group and only a 25% reduction in the placebo group.
Emicizumab
Emicizumab (Hemlibra, Emicizumab-KxWH, ACE910, RO5534262) is A bi-specific IgG4 antibody, targeting IXa and X, that was first approved by the FDA on November 16, 2017, for the prevention or reduction of bleeding frequency in hemophilia A patients containing clot factor VIII inhibitors. Prior to that, Emicizumab had been recognized as an orphan drug and breakthrough therapy by the FDA. Japan and Europe have been granted accelerated approval, while Japan has been granted orphan designation. Emicizumab was first discovered at home and abroad, developed together with Roche and marketed globally. The approval of this product is based on the clinical results of HAVEN1(NCT02622321) and interim results of HAVEN2(NCT02795767). According to the HAVEN1 study, hemophilia A patients 12 years and older with factor VIII inhibitors saw A significant 87% reduction in annual bleeding rates after receiving Hemlibra preventive therapy compared to those who did not receive prevention (2.9 vs 23.3). Interim results of the AVEN2 study showed that 87 per cent of children with hemophilia A who have inhibitors in their bodies under 12 years of age did not bleed after receiving Hemlibra prevention. In a patient-specific analysis of 13 pediatric patients enrolled in NIS, Hemlibra prophylactic treatment reduced bleeding rates by 99% compared with patients treated with bypassing agents (BPA).
Monoclonal antibodies under European and American review
As of December 1, 2017, a total of nine monoclonal antibody products have been reviewed in Europe and the United States. With the exception of Mogamulizumab, which was approved in Japan in 2012, ibalizumab, Burosumab, Tildrakizumab, Caplacizumab, Erenumab, Fremanezumab, Galcanezumab and Romosozumab have not been approved in any country. The products are expected to be available in 2018, with Burosumab and ibalizumab expected to be approved for sale in January or February.
Brodalumab (Siliq, Lumicef, Kyntheum, AMG-827) is a human IgG2 antibody, targeting the interleukin-17 receptor (IL-17RA). It can block the inflammatory signal of IL-17A through IL-17RA, and promote the inflammatory cytokines IL-17F and IL-17C. Brodalumab was first approved in Japan on July 4, 2016 for Lumicef, which includes erythrodermic psoriasis, pustular psoriasis, psoriatic arthritis and psoriasis vulgaris. In February 2017 and July 2017, Brodalumab was approved in the United States and Europe, respectively, for the treatment of moderate to severe plaque psoriasis in adults who do not respond to systemic therapy or light therapy (UV therapy). Safety and efficacy of Brodalumab was studied in three placebo-controlled phase III clinical trials, amagine-1 (NCT01708590), Amagine-2 (NCT01708603) and Amagine-3 (NCT01708603), involving a total of 4,373 patients. The primary end point was the proportion of patients with psoriasis at week 12 who improved by more than 75% in area with severity (PASI75) and whose physician's static overall assessment (sPGA) score (0/1) decreased by at least 2 points from baseline. Amagine-1 results showed that 83 per cent of Brodalumab 210mg to PASI75, 60 per cent of the 140mg group and 3 per cent of the placebo group. The sPGA scores were also higher than the placebo group at 76%, 54% and 1%, respectively. Amagine-2 and Amagine-3 test data showed that the improvement of clinical symptoms of moderate and severe psoriasis was significantly better than that of placebo group. In amagine-2, the PASI compliance rate was 86% in 210mg group, 67% in 140mg group, and 8% in placebo group, 86% in Amagine-3, 69% and 6% in amagine-3, respectively. The sPGA compliance rate was also higher in the treatment group, 79% in 210mg amagine-2, 58% in 140mg group, and 4% in placebo group. In Amagine-3, 80%, 60% and 4%, respectively.
Avelumab
Avelumab (Bavencio, MSB0010718C), a human IgG1 monoclonal antibody targeting PD-L1, was first approved on May 23, 2017. The indications approved were metastatic Merkel-cell carcinoma in adults and children over 12 years of age. The indication was also approved in Europe in September 2017. Avelumab was approved based on data from the Javelin Merkel 200 study (NCT02155647), A key phase II clinical trial in which Part A patients underwent chemotherapy prior to initial treatment with Avelumab in Part B. At a dose of 10mg/kg, intravenous injection is given fortnightly. In Part A, 88 patients who had undergone prior chemotherapy were treated with this product and their conditions were controlled. The median follow-up period was 10.4 months, and the objective remission rate was 31.8%, among which 8% had complete remission and 20% had partial remission. In Part B, 39 patients were treated for the first time with this product, and data on the median remission time of the primary endpoint is expected to be available in September 2019. Avelumab has previously been granted orphan status for Merckle cell cancer in Europe and Australia, as well as FDA breakthrough therapy designation. On May 9, 2017, THE FDA granted Avelumab a priority review of indications for locally advanced or metastatic urothelial carcinoma that has progressed after platinum chemotherapy. In addition, four phase III clinical trials of the product for non-small cell lung cancer, renal cell cancer, ovarian cancer and gastric cancer will be completed in 2018 and have been awarded orphan drug status for gastric cancer in both the United States and Europe.
Dupilumab
Dupilumab (Dupixent, REGN668/SAR231893), an IL4R-targeted IgG4 monoclonal antibody, was approved by the FDA on April 28, 2017, and by THE EMA on September 28, 2017, respectively, for the treatment of atopic dermatitis. Dupixent's approval was based on LIBERTY, an atopic dermatitis study that included three clinical trials of SOLO1, SOLO2 and CHRONOS. In SOLO1 and SOLO2, patients were treated with either 300mg Dupixent or placebo once a week, or with 300mg Dupixent every two weeks, respectively, and switched to placebo. The efficacy of Dupixent combined with corticosteroid was studied by CHRONOS test with placebo as control. The primary end point of the trial was the proportion of patients with a reduction of more than two points from baseline in the investigator's Overall Evaluation (IGA) score (0/1) at week 16. The results showed that, after 16 weeks of Dupixent monotherapy, the IGA compliance was significantly higher than that of the placebo group, with 37% of SOLO1 and 36% of SOLO2. Similarly, by CHRONOS, the dupilumab+ corticosteroid combination treatment was 39% and the placebo + steroid combination treatment was 12%. Because of its outstanding efficacy, this product has been recognized by FDA as a breakthrough therapy for atopic dermatitis. In addition to atopic dermatitis, Dupilumab is currently in phase III clinical development for indications for asthma and nasal polyps. In September 2017, Sanofi/Regeneration announced that dupilumab's LIBERTY ASTHMA QUEST (NCT02414854) trial for uncontrolled persistent ASTHMA had reached its therapeutic endpoint. Two other clinical trials on nasal polyp treatment (NCT02912468, NCT02898454) will also reach their endpoints in 2018. More encouragingly, this product can also be used in the treatment of eosinophilic esophagitis, and the current research is in the clinical phase II, and has been recognized as an orphan drug by FDA.
Ocrelizumab
Ocrelizumab (Ocrevus) is a humanized IgG1 monoclonal antibody that targets Cd-20-positive B cells. These B cells play a role in myelin damage and multiple sclerosis by producing pro-inflammatory cytokines, secreting autoantibodies, and activating pro-inflammatory T cells. Ocrevus destroys CD20 positive B cells in three main ways: complement dependent cytotoxicity and antibody-dependent cell-mediated cytotoxicity, and by binding to CD20 on target cells to apoptosis. This monoclonal antibody has been studied in both internal rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE), but the expected results are not satisfactory. Ocrevus received FDA approval on March 29 for relapsing and relapsing multiple sclerosis as well as primary progressive multiple sclerosis, following the indication's FDA breakthrough therapy designation, fast-track and priority review. Two trials, OPERAI (NCT01247324) and OPERAII (NCT01412333), respectively, assessed the safety and efficacy of Ocrevus in the treatment of multiple sclerosis with remission, and showed significantly lower recurrence rates in the Ocrevus group than in the beta interferon group, with OPERAI 46% lower and OPERAII 57% lower. In primary progressive multiple sclerosis, results from the ORATORIO trial showed that Ocrevus significantly reduced clinical disability progression at 12 weeks (primary endpoint) compared with placebo (32.9% vs 39.3%). In addition, MRI showed a 3.4 percent decrease in the overall volume of encephalopathy in patients treated with Ocrevus, compared with a 7.4 percent increase in placebo.
Durvalumab
Durvalumab (Imfinzi, MEDI4736) is a human IgG1 antibody that directly activates PD-L1 to block the association between PD-L1 and its receptor (PD-1, CD80). Durvalumab, designed to block the interaction between cytotoxic effects and PD-L1 positive immune cells, was approved by the FDA on May 1, 2017 for accelerated use in locally advanced or metastatic uEPITHELIAL carcinoma that is exacerbated by platinum chemotherapy. This is a combination of priority review and breakthrough therapy, based on a phase 1/2 clinical trial of 182 patients (NCT01693562). Durvalumab was given intravenously every two weeks at a dose of 10mg/kg for 12 months, and the objective remission rate was 27.4% in the subgroup with high EXPRESSION of PD-L1 and 4.1% in the subgroup with low expression of PD-L1. In addition, the NDA for locally advanced or inoperable non-small cell lung cancer (NSCLC) after platinum chemotherapy has entered the FDA review stage, having previously obtained FDA priority review and breakthrough therapy recognition. At the same time, an application for marketing permission (MAA) for this indication has been accepted by EMA, and data from the phase III PACIFI clinical trial (NCT02125461) has also been submitted to EMA. Clinical results showed that for NSCLC progressing after chemotherapy, the median progression-free survival was 16.8 months, while the placebo was only 5.6 months. In addition to the above two indications, Durvalumab has been granted fast-track status by FDA for the treatment of pD-L1-positive metastatic head and neck cancer. Phase III clinical trials EAGLE (NCT02369874) and KESTREL (NCT02551159) are expected to reach their endpoints in February and March 2018, respectively.
Sarilumab
Sarilumab (Kevzara SAR153191, REGN88) is a kind of IL - 6 r as targets of IgG1 sheet resistance, in February 2017, approved, for the first time in Canada in May 2017 and July 2017 respectively and cleared by the FDA and EMA for one or more biological products or biological products regulatory anti-rheumatism disease drugs (DMARDs) inadequate response or intolerance of the relation between active rheumatoid arthritis. Sarilumab's approval was based on a global multicenter Sari-RA clinical trial involving MOBILITY (NCT01061736), TARGET (NCT01709578) and MONARCH (NCT02332590). Sarilumab was used in combination with methotrexate or other DMARD in both the MOBILITY and TARGET trials. Results of the MOBILITY study showed that ACR20 at 24 weeks was 66% in the 200mg sarilumab treatment group, 58% in the 150mg group, and only 33% in the placebo group. Sarilumab also improved patients' physiological function. Haq-di score decreased by 0.58 in the 200mg Sarilumab treatment group, 0.54 in the 150mg group, and 0.30 in the placebo group. TARGET showed similar results: 61% of ACR20 in the DMARD+200mg Sarilumab group, 56% in the 150mg group, and 34% in the placebo group. In terms of physiological function improvement, haq-DI score decreased by 0.50 in the DMARD+200mg Sarilumab treatment group and 0.49 in the 150mg group, while only 0.29 in the placebo group. The MONARCH test assessed the safety and effectiveness of Sarilumab against adamumab. At 24 weeks, the disease activity score (DAS28-ESR) for 28 joints calculated using ERYTHROcyte sedimentation rate decreased by 3.28 from baseline, while the adamumab group decreased by only 2.20, which was also the primary endpoint.
Guselkumab
Guselkumab (Tremfya) is a human IgG1, which blocks the P19 subunit of IL-23, thereby limiting inflammatory response. Il-23 is a cytokine involved in Th17 cell differentiation and maintenance, producing pro-inflammatory cytokines IL-17, IL-22 and TNF through T cell subsets. On July 13, 2017, Guselkumab received FDA approval for moderate to severe plaque psoriasis for systemic therapy (injection or oral treatment) or phototherapy (ultraviolet treatment), and in November 2017, guselkumab was reapproved by EMA for the same indications as in the United States. Guselkumab was granted based on the results of three clinical trials, VOYAGE1 (NCT02207231), VOYAGE2 (NCT02207244) and NAVIGATE (NCT02203032). VOYAGE1 and VOYAGE2 evaluated the efficacy and safety of Tremfya relative to placebo and altamumab. The results showed that after 16 weeks of guselkumab treatment, the IGA compliance rate was far higher than that of the placebo group (85.1% vs 6.9%). In addition, 73.3% and 70.0% of the patients in the Tremfya treatment group achieved PASI90 remission, while 49.7% and 46.8% of the altamumab treatment group achieved relief, respectively. The NAVIGATE test evaluates the efficacy and safety of Tremfya versus Stelara. Patients receive Ustekinumab at week 0 and 4, and patients with inadequate response are given ustekinumab or guselkumab again at week 16. The primary end point is the proportion of patients whose IGA score has decreased by a minimum of 2 points at week 28 from baseline. The results showed that after treatment with ustekinumab or guselkumab, the IGA score reaching rates were 14.3% and 31.1% at 28 weeks, and 17.3% and 36.3% at 32 weeks, respectively. The data indicate that Tremfya still has significant therapeutic effect in patients with inadequate response to previous treatment with Stelara.
Inotuzumab ozogamicin
Inotuzumab ozogamicin (Besponsa, CMC-544) is a conjugate of humanized IgG4 antibody against CD22 and the cytotoxic drug Kazamycin. Besponsa was first approved in Europe on June 30, 2017 and again approved by THE FDA on August 17, 2017 for both adult relapsed or refractory B-cell precursor acute lymphoblastic leukemia (ALL). Prior to this, Besponsa has been approved as an orphan drug in Europe and the United States, as well as FDA's ALL Breakthrough Therapy designation. Besponsa was approved on the basis of a phase III clinical trial called INOVATE ALL (NCT01564784), which is controlled with existing standard therapies. Patients received three doses of Besponsa or standard chemotherapy on day 1 (0.8mg/m2), day 8 (0.5mg/m2), and day 15 (0.5mg/m2). The initial course is 21 days, and then each course is 28 days, leading to 6 courses. The results showed that the Besponsa group had a complete response rate of 80.7% and a median total survival time of 7.7 months, while the standard chemotherapy group had a complete response rate of 29.4% and a median total survival time of 6.7 months.
Benralizumab
Benralizumab (Fasenra, Medi-563) is a defucified IgG1 monoclonal antibody targeting the subunit of IL-5R. On November 14, 2017, THE FDA approved this product for additional maintenance therapy in patients with severe asthma aged 12 years and older with eosinophilic phenotypes, and on November 10, 2017, EMA's expert committee also gave a positive opinion on the release of this product. The safety and effectiveness of this product was evaluated in the WINDWARD study, which included 6 phase III clinical trials, including SIROCCO, CALIMA, ZONDA, BISE, BORA and GREGALE. Two randomized, double-blind, parallel, placebo-controlled clinical trials, SIROCCO(NCT01928771) and CALIMA (NCT01914757), evaluated the long-term safety of benralizumab. Patients were given a fixed subcutaneous injection of 30mg for 56 weeks, and the results of the study were published in 2016. In the 28-week ZONDA(NCT02075255) trial, patients with moderate to severe asthma were treated with either Benralizumab 30mg or placebo once every four or eight weeks on oral corticosteroids. The results showed a 75% reduction in corticosteroids in the Benralizumab group and only a 25% reduction in the placebo group.
Emicizumab
Emicizumab (Hemlibra, Emicizumab-KxWH, ACE910, RO5534262) is A bi-specific IgG4 antibody, targeting IXa and X, that was first approved by the FDA on November 16, 2017, for the prevention or reduction of bleeding frequency in hemophilia A patients containing clot factor VIII inhibitors. Prior to that, Emicizumab had been recognized as an orphan drug and breakthrough therapy by the FDA. Japan and Europe have been granted accelerated approval, while Japan has been granted orphan designation. Emicizumab was first discovered at home and abroad, developed together with Roche and marketed globally. The approval of this product is based on the clinical results of HAVEN1(NCT02622321) and interim results of HAVEN2(NCT02795767). According to the HAVEN1 study, hemophilia A patients 12 years and older with factor VIII inhibitors saw A significant 87% reduction in annual bleeding rates after receiving Hemlibra preventive therapy compared to those who did not receive prevention (2.9 vs 23.3). Interim results of the AVEN2 study showed that 87 per cent of children with hemophilia A who have inhibitors in their bodies under 12 years of age did not bleed after receiving Hemlibra prevention. In a patient-specific analysis of 13 pediatric patients enrolled in NIS, Hemlibra prophylactic treatment reduced bleeding rates by 99% compared with patients treated with bypassing agents (BPA).
Monoclonal antibodies under European and American review
As of December 1, 2017, a total of nine monoclonal antibody products have been reviewed in Europe and the United States. With the exception of Mogamulizumab, which was approved in Japan in 2012, ibalizumab, Burosumab, Tildrakizumab, Caplacizumab, Erenumab, Fremanezumab, Galcanezumab and Romosozumab have not been approved in any country. The products are expected to be available in 2018, with Burosumab and ibalizumab expected to be approved for sale in January or February.
Monoclonal antibodies under EMA/FDA review in 2017

Ibalizumab
Ibalizumab, an IgG4 monoclonal antibody targeting CD4, is currently under FDA review and has been applied for the indication of multiple antiviral drugs for the treatment of drug-resistant HIV infection. This is a combination of orphan drug qualification and breakthrough therapy, and due to the FDA's priority review, PDUFA expires on January 3, 2018. The BLA was supported by a phase III clinical trial called TMB-301(NCT02475629), which investigated the safety and efficacy of Ibalizumab in an open-label manner. In October 2017, Thera Technologies Inc announced that the TMB301 study had been completed and that it would continue to expand the research project TMB-311(NCT02707861). Twenty-seven patients who completed the 24-week TMB-301 trial were enrolled in the TMB-311 study and received ibalizumab 800mg every two weeks for 48 weeks. In the TMB-311 trial, 15 patients with no detectable viral load at 24 weeks were maintained for 48 weeks. Of the remaining patients whose viral load was detectable at 24 weeks, 17 (63%) dropped below 200 copies /ml at 48 weeks.
Burosumab
Burosumab (KRN23) is a human IgG1 monoclonal antibody targeting fibroblast growth factor 23(FGF23). Burosumab was discovered by Kirin Of Japan and applied for indications for familial hypophosphatemia rickets (XLH). FGF23 is a hormone associated with renal phosphorus secretion control and active vitamin D production. The disease is characterized by skeletal muscle defects due to excess LEVELS of FGF23 and tumor-induced osteomalacia. Burosumab's marketing application has been submitted to both FDA and EMA. In the United States, Burosumab has received recognition and priority review for XLH breakthrough Therapy. In Europe, Burosumab has received positive comments from THE CHMP until April 17, 2018. In a randomized, double-blind, placebo-controlled phase III clinical trial (NCT02526160), Burosumab significantly increased blood phosphorus levels in XLH patients, reaching the primary endpoint of treatment. In this trial, 35 XLH patients were randomly assigned to 1mg/kg burosumab or placebo once every 4 weeks for 24 weeks. Results showed that 94% of patients in the burosumab group (n=64) had blood phosphorus levels above the lower limit and maintained blood phosphorus levels within the normal range for 24 weeks, compared with only 8% of patients in the placebo group. Another clinical trial of Burosumab in combination with oral phosphorus/active vitamin D for pediatric XLH (NCT02915705) will be completed in July 2018.
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222) is a human IgG1 monoclonal antibody targeting IL-23P19. Marketing applications for moderate to severe plaque psoriasis have been submitted to FDA and EMA. Data supporting the BLA came out of two phase III clinical trials, in which more than 1,800 patients were included in the study, some of which ended up being treated for up to 3.5 years. A 52-week, placebo-controlled, parallel-designed reSURFACE2 trial (NCT01729754) compared the safety and tolerability of Tildrakizumab (100/200mg) against etanercep. ReSURFACE1 (NCT017223310) was designed similarly to reSURFACE2, but it added active drug control. During the 12 weeks of the trial, 66% of patients in the 200mg tildrakizumab group and 61% in the 100mg group ended up with PASI75, compared with 48% in the enaciprit group and 6% in the placebo group. The PGA compliance rates were 59% in both tildrakizumab 200mg and 100mg, compared with 48% in the etanercept group and 4% in the placebo group. The reSURFACE1 experiment ended up with similar results.
Caplacizumab
Caplacizumab (ALX-0081) is a nanoscale antibody targeting von Willebrand factor for which an indication has been proposed as acquired thrombotic thrombocytopenic purpura (aTTP). This is a rare and severe form of disseminated thrombotic microangiopathy, characterized by microangiopathic hemolytic anemia, decreased platelet aggregation consumption, and damage to organs (e.g., kidneys, central nervous system, etc.) caused by microthrombus formation. Caplacizumab has previously been recognized as an aTTP orphan drug by FDA and EMA, respectively, with FDA support for a fast-track review channel. In February 2017, Ablynx announced that MAA had been submitted to EMA, that HERCULES data from phase III clinical trials of aTTP had been obtained, and that the safety and efficacy of the product had been confirmed. A total of 145 patients with acute aTTP were recruited. Patients were randomized 1:1 to the Caplacizumab or placebo group, and all were given standard care, namely daily plasma exchange and immunosuppressive agents. Patients were intravenously injected with caplacizumab or placebo 10mg on the first day after plasmapheresis, followed by daily subcutaneous injection of Caplacizumab or placebo for 30 consecutive days. An additional treatment period of 7-28 days was added to the 30 days, depending on the patient's response, with the primary endpoint being the time required for platelet count response. Results show that a caplacizumab platelet count than placebo response time of patients treated statistically significantly reduced, the probability of response to platelet count in patients with increased by 50%, mortality, recurrence aTTP patient during test, a more comprehensive by 74%, the incidence of thrombotic events throughout the study period of aTTP recurrence rate was 67%. Patients who have completed the HERCULES trial have been enrolled in the three-year follow-up trial (NCT02878603), which is ongoing.
Erenumab
Erenumab (Aimovig, AMG334) is a human IgG2 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), which is thought to be associated with the progression of sensory neuron sensitization and impairment. The proposed indication for migraine has been submitted to FDA and EMA. The product is jointly developed by Amgen and Novartis, which owns the rights to operate the product in the United States. PDUFA expires on May 17, 2018. The rights to operate the product in Europe and other regions except Japan are owned by Novartis. The data supporting BLA were the results of four phase II or III clinical trials. More than 2,600 patients enrolled in clinical trials with more than four headaches per month demonstrated that Erenumab significantly reduced the number of headache days per month in patients compared with placebo, and significantly reduced the number of medication sessions for headache disability or acute episodes. In a clinical trial of NCT02456740, 955 participants had an average of 8.3 days of migraine per month. After four to six months of the trial, the 70mg Erenumab group reduced migraine headaches by 3.2 days, the 140mg Erenumab group by 3.7 days, and the placebo group by 1.8 days. The number of migraine days decreased by 50% or more in 43.4% in the 70mg Erenumab group and 50.0% in the 140mg Erenumab group, compared with 26.6% in the placebo group. Functional impairment scores decreased by 4.2 points in the 70mg Erenumab group, 4.8 points in the 140mg Erenumab group, and 2.4 points in the placebo group. Daily activity scores improved 5.5 points in the 70mg Erenumab group, 5.9 points in the 140mg Erenumab group and 3.3 points in the placebo group.
Fremanezumab
Fremanezumab (TEV-48125) is an IgG2 monoclonal antibody that targets calcitonin gene-related peptide (CGRP) and is also indicated for migraine. The data supporting BLA included data from phase III clinical trials, including the HALO study program, which included more than 2,000 patients with recurrent or chronic migraine and had both primary and secondary endpoints. Patients enrolled in the HALO study were randomly assigned to one of three groups: one group was given subcutaneous fremanezumab 225mg/ month for three months, the other group was given fremanezumab 675mg initially, followed by a placebo for two months, and a third group was given a corresponding placebo. The treatment end point of the trial was 12 weeks, relative to baseline changes in the number of headache days per month. The results showed a significant decrease of 41.6% (-3.7 vs 2.2 days) in the number of migraine days per month compared with baseline, and a decrease of 3.4 days or 37.0% in the number of migraine days per month compared with the quarterly regimen. Chronic migraine patients enrolled in the HALO study were also randomly assigned to one of three groups: one group was given fremanezumab 225mg/ month subcutaneously for three months, another group was given Fremanezumab 675mg initially, followed by a placebo for two months, and a third group was given a placebo. The results showed that the number of migraine days per month decreased by 2.5 days over 12 weeks compared to placebo, 4.6 days per month for the monthly regimen, and 4.3 days per month for the quarterly regimen.
Galcanezumab
Galcanezumab (LY2951742) is an IgG4 monoclonal antibody that targets calcitonin gene-related peptide (CGRP) and is also indicated for migraine. The clinical data supporting the BLA were derived from 3 phase III clinical trials evolutions -1, evolutions -2, and this. In EVOLVE-1, the number of headache days per month decreased by 50% in the 120mg Galcanezumab group, 60.9% in the 240mg Group, 38.6% in the placebo group, 38.8% in the 120mg Galcanezumab group, 38.5% in the 240mg group, and 19.3% in the placebo group. The number of headache days per month decreased by 100% relative to baseline in the 120mg Galcanezumab group was 15.6%, 14.6% in the 240mg group, and 6.2% in the placebo group. Similar results were obtained for the evolutions -2 trial. In a placebo-controlled trial, patients with chronic migraine were given 120 or 240mg of galcanezumab for more than three months, and their average number of headache days per month was significantly lower than that of the control group. In addition, the product is currently undergoing a placebo-controlled phase III clinical study of chronic cluster headache (NCT02438826). Patients were given a monthly subcutaneous injection of Galcanezumab for 12 months. The primary outcome measure was an improvement in weekly cluster headache frequency relative to baseline. The clinical trial has enrolled 162 subjects and results are expected in March 2018. Previously, the indication had been granted fast-track FDA approval, which is expected to speed the indications for cluster headache to market.
Romosozumab
Romosozumab (EVENITYTM, AMG785) is a humanized IgG2 monoclonal antibody, targeting osteocalcin. Romosozumab's BLA has been submitted to FDA in July 2016 for indication as osteoporosis. Data supporting the BLA included results from the Placebo-controlled FRAME(NCT01575834) Phase III clinical study. Subjects received 210mg of romosozumab subcutaneously once a month for 12 months, and 60mg of romosozumab subcutaneously semiannually for 24 months. Then phase 3 trials ARCH (NCT01631214), according to the results of a monthly subcutaneous injection of this product is 210 mg compared with weekly oral alendronate phosphonic acid sodium, serious cardiovascular adverse events increased significantly (2.5% vs 1.9%), in July 2017, announced that the FDA fully return of amgen, asking for the safety of the ARCH and the third phase of clinical trial data BRIDGE (NCT02186171) data, to assess the efficacy of male patients with osteoporosis drug safety.
Mogamulizumab
Mogamulizumab (KW-0761, Poteligeo) is a deglycosylated humanized IgG1 monoclonal antibody that targets CC chemokine receptor 4(CCR4) expressed on tumor cells in patients with cutaneous T cell lymphoma. This product has been approved by Japan PMDA for the treatment of recurrent or refractory CCR4-positive adult T-cell leukemia (ATL) in March 2012, cCR4-positive peripheral T-cell lymphoma and cutaneous T-cell lymphoma in March 2014, and cCR4-positive adult T-cell leukemia without chemotherapy in December 2014. In July 2017, Kirin of Japan announced that mogamulizumab's application for marketing as a treatment for T-cell lymphoma that has recurred after systemic treatment with at least one drug is under EMA review. The data supporting MAA were from a randomized, open-label, multicentre phase III clinical trial, MAVORIC (NCT01728805), which assessed the safety and effectiveness of mogamulizumab using vorinostat as the control. 372 refractory CTCL patients were randomized to vorinostat or mogamulizumab. In April 2017, qilin announced that the MAVORIC trial reached the established clinical endpoint of PFS (254 days vs169 days), and in August 2017, mogamulizumab was identified by the FDA as a groundbreaking therapy for mycosis granuloma and cezary syndrome.
The monoclonal antibody will file for listing in 2018
Data on key clinical endpoints have been obtained or will be available in the near future, and 12 monoclonal antibodies are expected to submit BLAS in 2018. They are Lanadelumab, Crizanlizumab, Ravulizumab, Eptinezumab, Risankizumab, Satralizumab, Brolucizumab, PRO140, Sacituzumab Govitecan, Moxetumomab Pasudotox, Cemiplimab and ublituximab. The 12 drugs are also expected to be on the market in late 2018 or 2019, with the first eight being generic and the last four anticancer drugs, according to the FDA's review cycle.
Ibalizumab, an IgG4 monoclonal antibody targeting CD4, is currently under FDA review and has been applied for the indication of multiple antiviral drugs for the treatment of drug-resistant HIV infection. This is a combination of orphan drug qualification and breakthrough therapy, and due to the FDA's priority review, PDUFA expires on January 3, 2018. The BLA was supported by a phase III clinical trial called TMB-301(NCT02475629), which investigated the safety and efficacy of Ibalizumab in an open-label manner. In October 2017, Thera Technologies Inc announced that the TMB301 study had been completed and that it would continue to expand the research project TMB-311(NCT02707861). Twenty-seven patients who completed the 24-week TMB-301 trial were enrolled in the TMB-311 study and received ibalizumab 800mg every two weeks for 48 weeks. In the TMB-311 trial, 15 patients with no detectable viral load at 24 weeks were maintained for 48 weeks. Of the remaining patients whose viral load was detectable at 24 weeks, 17 (63%) dropped below 200 copies /ml at 48 weeks.
Burosumab
Burosumab (KRN23) is a human IgG1 monoclonal antibody targeting fibroblast growth factor 23(FGF23). Burosumab was discovered by Kirin Of Japan and applied for indications for familial hypophosphatemia rickets (XLH). FGF23 is a hormone associated with renal phosphorus secretion control and active vitamin D production. The disease is characterized by skeletal muscle defects due to excess LEVELS of FGF23 and tumor-induced osteomalacia. Burosumab's marketing application has been submitted to both FDA and EMA. In the United States, Burosumab has received recognition and priority review for XLH breakthrough Therapy. In Europe, Burosumab has received positive comments from THE CHMP until April 17, 2018. In a randomized, double-blind, placebo-controlled phase III clinical trial (NCT02526160), Burosumab significantly increased blood phosphorus levels in XLH patients, reaching the primary endpoint of treatment. In this trial, 35 XLH patients were randomly assigned to 1mg/kg burosumab or placebo once every 4 weeks for 24 weeks. Results showed that 94% of patients in the burosumab group (n=64) had blood phosphorus levels above the lower limit and maintained blood phosphorus levels within the normal range for 24 weeks, compared with only 8% of patients in the placebo group. Another clinical trial of Burosumab in combination with oral phosphorus/active vitamin D for pediatric XLH (NCT02915705) will be completed in July 2018.
Tildrakizumab
Tildrakizumab (SCH900222/MK-3222) is a human IgG1 monoclonal antibody targeting IL-23P19. Marketing applications for moderate to severe plaque psoriasis have been submitted to FDA and EMA. Data supporting the BLA came out of two phase III clinical trials, in which more than 1,800 patients were included in the study, some of which ended up being treated for up to 3.5 years. A 52-week, placebo-controlled, parallel-designed reSURFACE2 trial (NCT01729754) compared the safety and tolerability of Tildrakizumab (100/200mg) against etanercep. ReSURFACE1 (NCT017223310) was designed similarly to reSURFACE2, but it added active drug control. During the 12 weeks of the trial, 66% of patients in the 200mg tildrakizumab group and 61% in the 100mg group ended up with PASI75, compared with 48% in the enaciprit group and 6% in the placebo group. The PGA compliance rates were 59% in both tildrakizumab 200mg and 100mg, compared with 48% in the etanercept group and 4% in the placebo group. The reSURFACE1 experiment ended up with similar results.
Caplacizumab
Caplacizumab (ALX-0081) is a nanoscale antibody targeting von Willebrand factor for which an indication has been proposed as acquired thrombotic thrombocytopenic purpura (aTTP). This is a rare and severe form of disseminated thrombotic microangiopathy, characterized by microangiopathic hemolytic anemia, decreased platelet aggregation consumption, and damage to organs (e.g., kidneys, central nervous system, etc.) caused by microthrombus formation. Caplacizumab has previously been recognized as an aTTP orphan drug by FDA and EMA, respectively, with FDA support for a fast-track review channel. In February 2017, Ablynx announced that MAA had been submitted to EMA, that HERCULES data from phase III clinical trials of aTTP had been obtained, and that the safety and efficacy of the product had been confirmed. A total of 145 patients with acute aTTP were recruited. Patients were randomized 1:1 to the Caplacizumab or placebo group, and all were given standard care, namely daily plasma exchange and immunosuppressive agents. Patients were intravenously injected with caplacizumab or placebo 10mg on the first day after plasmapheresis, followed by daily subcutaneous injection of Caplacizumab or placebo for 30 consecutive days. An additional treatment period of 7-28 days was added to the 30 days, depending on the patient's response, with the primary endpoint being the time required for platelet count response. Results show that a caplacizumab platelet count than placebo response time of patients treated statistically significantly reduced, the probability of response to platelet count in patients with increased by 50%, mortality, recurrence aTTP patient during test, a more comprehensive by 74%, the incidence of thrombotic events throughout the study period of aTTP recurrence rate was 67%. Patients who have completed the HERCULES trial have been enrolled in the three-year follow-up trial (NCT02878603), which is ongoing.
Erenumab
Erenumab (Aimovig, AMG334) is a human IgG2 monoclonal antibody that targets calcitonin gene-related peptide (CGRP), which is thought to be associated with the progression of sensory neuron sensitization and impairment. The proposed indication for migraine has been submitted to FDA and EMA. The product is jointly developed by Amgen and Novartis, which owns the rights to operate the product in the United States. PDUFA expires on May 17, 2018. The rights to operate the product in Europe and other regions except Japan are owned by Novartis. The data supporting BLA were the results of four phase II or III clinical trials. More than 2,600 patients enrolled in clinical trials with more than four headaches per month demonstrated that Erenumab significantly reduced the number of headache days per month in patients compared with placebo, and significantly reduced the number of medication sessions for headache disability or acute episodes. In a clinical trial of NCT02456740, 955 participants had an average of 8.3 days of migraine per month. After four to six months of the trial, the 70mg Erenumab group reduced migraine headaches by 3.2 days, the 140mg Erenumab group by 3.7 days, and the placebo group by 1.8 days. The number of migraine days decreased by 50% or more in 43.4% in the 70mg Erenumab group and 50.0% in the 140mg Erenumab group, compared with 26.6% in the placebo group. Functional impairment scores decreased by 4.2 points in the 70mg Erenumab group, 4.8 points in the 140mg Erenumab group, and 2.4 points in the placebo group. Daily activity scores improved 5.5 points in the 70mg Erenumab group, 5.9 points in the 140mg Erenumab group and 3.3 points in the placebo group.
Fremanezumab
Fremanezumab (TEV-48125) is an IgG2 monoclonal antibody that targets calcitonin gene-related peptide (CGRP) and is also indicated for migraine. The data supporting BLA included data from phase III clinical trials, including the HALO study program, which included more than 2,000 patients with recurrent or chronic migraine and had both primary and secondary endpoints. Patients enrolled in the HALO study were randomly assigned to one of three groups: one group was given subcutaneous fremanezumab 225mg/ month for three months, the other group was given fremanezumab 675mg initially, followed by a placebo for two months, and a third group was given a corresponding placebo. The treatment end point of the trial was 12 weeks, relative to baseline changes in the number of headache days per month. The results showed a significant decrease of 41.6% (-3.7 vs 2.2 days) in the number of migraine days per month compared with baseline, and a decrease of 3.4 days or 37.0% in the number of migraine days per month compared with the quarterly regimen. Chronic migraine patients enrolled in the HALO study were also randomly assigned to one of three groups: one group was given fremanezumab 225mg/ month subcutaneously for three months, another group was given Fremanezumab 675mg initially, followed by a placebo for two months, and a third group was given a placebo. The results showed that the number of migraine days per month decreased by 2.5 days over 12 weeks compared to placebo, 4.6 days per month for the monthly regimen, and 4.3 days per month for the quarterly regimen.
Galcanezumab
Galcanezumab (LY2951742) is an IgG4 monoclonal antibody that targets calcitonin gene-related peptide (CGRP) and is also indicated for migraine. The clinical data supporting the BLA were derived from 3 phase III clinical trials evolutions -1, evolutions -2, and this. In EVOLVE-1, the number of headache days per month decreased by 50% in the 120mg Galcanezumab group, 60.9% in the 240mg Group, 38.6% in the placebo group, 38.8% in the 120mg Galcanezumab group, 38.5% in the 240mg group, and 19.3% in the placebo group. The number of headache days per month decreased by 100% relative to baseline in the 120mg Galcanezumab group was 15.6%, 14.6% in the 240mg group, and 6.2% in the placebo group. Similar results were obtained for the evolutions -2 trial. In a placebo-controlled trial, patients with chronic migraine were given 120 or 240mg of galcanezumab for more than three months, and their average number of headache days per month was significantly lower than that of the control group. In addition, the product is currently undergoing a placebo-controlled phase III clinical study of chronic cluster headache (NCT02438826). Patients were given a monthly subcutaneous injection of Galcanezumab for 12 months. The primary outcome measure was an improvement in weekly cluster headache frequency relative to baseline. The clinical trial has enrolled 162 subjects and results are expected in March 2018. Previously, the indication had been granted fast-track FDA approval, which is expected to speed the indications for cluster headache to market.
Romosozumab
Romosozumab (EVENITYTM, AMG785) is a humanized IgG2 monoclonal antibody, targeting osteocalcin. Romosozumab's BLA has been submitted to FDA in July 2016 for indication as osteoporosis. Data supporting the BLA included results from the Placebo-controlled FRAME(NCT01575834) Phase III clinical study. Subjects received 210mg of romosozumab subcutaneously once a month for 12 months, and 60mg of romosozumab subcutaneously semiannually for 24 months. Then phase 3 trials ARCH (NCT01631214), according to the results of a monthly subcutaneous injection of this product is 210 mg compared with weekly oral alendronate phosphonic acid sodium, serious cardiovascular adverse events increased significantly (2.5% vs 1.9%), in July 2017, announced that the FDA fully return of amgen, asking for the safety of the ARCH and the third phase of clinical trial data BRIDGE (NCT02186171) data, to assess the efficacy of male patients with osteoporosis drug safety.
Mogamulizumab
Mogamulizumab (KW-0761, Poteligeo) is a deglycosylated humanized IgG1 monoclonal antibody that targets CC chemokine receptor 4(CCR4) expressed on tumor cells in patients with cutaneous T cell lymphoma. This product has been approved by Japan PMDA for the treatment of recurrent or refractory CCR4-positive adult T-cell leukemia (ATL) in March 2012, cCR4-positive peripheral T-cell lymphoma and cutaneous T-cell lymphoma in March 2014, and cCR4-positive adult T-cell leukemia without chemotherapy in December 2014. In July 2017, Kirin of Japan announced that mogamulizumab's application for marketing as a treatment for T-cell lymphoma that has recurred after systemic treatment with at least one drug is under EMA review. The data supporting MAA were from a randomized, open-label, multicentre phase III clinical trial, MAVORIC (NCT01728805), which assessed the safety and effectiveness of mogamulizumab using vorinostat as the control. 372 refractory CTCL patients were randomized to vorinostat or mogamulizumab. In April 2017, qilin announced that the MAVORIC trial reached the established clinical endpoint of PFS (254 days vs169 days), and in August 2017, mogamulizumab was identified by the FDA as a groundbreaking therapy for mycosis granuloma and cezary syndrome.
The monoclonal antibody will file for listing in 2018
Data on key clinical endpoints have been obtained or will be available in the near future, and 12 monoclonal antibodies are expected to submit BLAS in 2018. They are Lanadelumab, Crizanlizumab, Ravulizumab, Eptinezumab, Risankizumab, Satralizumab, Brolucizumab, PRO140, Sacituzumab Govitecan, Moxetumomab Pasudotox, Cemiplimab and ublituximab. The 12 drugs are also expected to be on the market in late 2018 or 2019, with the first eight being generic and the last four anticancer drugs, according to the FDA's review cycle.
Monoclonal antibodies are expected to file for listing in 2018
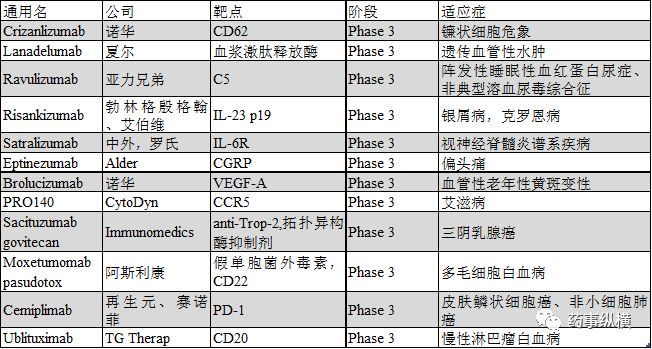
Lanadelumab
Lanadelumab (SHP643, DX-2930) is a human IgG1 monoclonal antibody that blocks bradykinin production by inhibiting plasma kinidin releasing enzyme. The indication for Lanadelumab in phase III was hereditary angioedema (HAE), a rare condition characterized by tissue edema following exposure. Lanadelumab has been granted orphan eligibility for HAE and FDA breakthrough Therapy for HAE in the U.S. and Europe. In May 2017, Shire reported data from a 26-week phase III clinical HELP trial (NCT02586805) that assessed the safety and efficacy of Lanadelumab in preventing the incidence of acute vascular edema in patients with HAE type I and II. Forty-nine patients were given either 300mg of lanadelumab every two weeks, 300mg of lanadelumab every four weeks, or 150mg of lanadelumab every four weeks or placebo. The results showed that the incidence of angioedema decreased by 87%, 73%, and 76%, respectively, compared with placebo in patients who were given Lanadelumab 300mg once every two weeks, 300mg once every four weeks, and 150mg lanadelumab 150mg once every four weeks, reaching the established primary and secondary endpoints. Another open-label phase III clinical long-term safety and efficacy study (NCT02741596) was conducted to assess the efficacy of Lanadelumab in preventing the development of acute vascular edema in patients with HAE I and II, with the end point expected in February 2018. Marketing applications for this product will be submitted to the FDA and EMA in early 2018.
Crizanlizumab
Crizanlizumab (SEG101) is a humanized monoclonal antibody that targets a platelet-based selective protein known as CD62. The indications studied are sickle cell pain crisis (SCPC). Sickle cell crisis is clinically manifested as chronic hemolytic anemia, prone to infection and recurrent pain crises leading to chronic ischemia and local damage to organs and tissues. In the placebo controlled phase II SUSTAIN test (NCT01895361), 198 patients aged 16 to 65 years were treated with Crizanlizumab every four weeks at a dose of 5mg/kg for 50 consecutive weeks. The median annual rate of SCPC decreased by 45.3%(1.63 vs 2.98), and the time of first OCCURRENCE of SCPC was 2.9 times longer than that of placebo (4.07 vs 1.38 months). The second occurrence of SCPC was 2.0 times longer than placebo (median 10.32 months and 5.09 months, respectively). Previously, Crizanlizumab has been certified as an ORPHAN SCPC in the United States and Europe, and novartis will submit the BLA in 2018 if the PK/PD data is consistent with the final manufacturing process.
Ravulizumab
Ravulizumab (ALXN1210), a humanized monoclonal antibody targeting complement 5(C5), is the next generation of Eculizumab, currently in the pre-registration stage. Its clinical trials included two phase III trials associated with paroxysmal haemoglobinuria (PNH) and two phase III trials associated with atypical hemolytic uremic syndrome (aHUS). These two (PNH) phase III clinical trials NCT02946463 and NCT03056040 are based on the results of a phase 1/2 clinical trial. Plasma lactate dehydrogenase (LDH) levels decreased rapidly and continuously after Ravulizumab in patients without complement inhibitor treatment, and FACIT fatigue scale scores were significantly improved. Phase 3 clinical trial NCT02946463 was designed to assess the safety and efficacy of Ravulizumab versus Eculizumab in adult PNH patients not treated with a complement inhibitor. In this trial, patients were given a loading dose on the first day based on body weight, and maintenance dose on the 15th day (40 to 60kg body weight: loading dose of 2400mg, maintenance dose of 3000mg every 8 weeks; 60 to 100kg body weight: 2700mg loading dose, 3300mg maintenance dose every 8 weeks; For patients with body weight greater than 100kg, the loading dose was 3000mg and the maintenance dose was 3600mg every 8 weeks), while eculizumab was administered according to the instructions. The expected clinical end point was a normal level of plasma lactate dehydrogenase (LDH) within 26 weeks. The trial enrolled 246 patients and will end in December 2017. Another phase 3 trial was designed to evaluate Ravulizumab in stable patients treated with Eculizumab for more than 6 months. Ravulizumab patients were given the same regimen as in the previous trial, while eculizumab patients were given a loading dose of Ravulizumab on Day 183 and a maintenance dose on day 197, with the primary end point being changes in plasma lactate dehydrogenase (LDH) levels at 26 weeks. The trial, which enrolled 197 patients, will end in March 2018. Alex is expected to report the results of the two trials in the second quarter of 2018 and submit the BLA. Ravulizumab has previously been recognized as an orphan drug for PNH in the United States and Europe. The single-arm phase III clinical trial associated with aHUS enrolled 55 patients and is expected to conclude in early 2018. Another phase III clinical trial involving children WITH aHUS, NCT03131219, is also underway and will end in December 2018.
Eptinezumab
Eptinezumab (ALD403) is an IgG1 monoclonal antibody targeting a calcitonin gene-related peptide (CGRP) that was developed for migraine. In June 2017, Alder BioPharma announced that the Phase III Clinical PROMISE1 trial (NCT02559895) was achieving a primary and secondary endpoint. The average number of headache days was 8.6 days/month for patients in the study who were given EPtinezumab 300mg, 100mg, 30mg or placebo, while the low-dose 30mg group was not included in the results. The results showed that a significant decrease in the number of headaches per month was observed between weeks 1 and 12, with a monthly headache of 4.3 days in the 300mg group, 3.9 days in the 100mg group, and 3.2 days in the placebo group. Nearly one-third of patients experienced a 75% or more decrease in the number of headache days per month at week 4-12, with an average of one-fifth of patients having no headache for at least one month within 1-6 months. Another Phase 3 clinical trial in PROMISE2 (NCT02974153) specifically designed for chronic migraine has enrolled 1,050 patients and will be completed in the first half of 2018, with Alder planning to file a marketing application in the second quarter of 2018.
Risankizumab
Risankizumab (ABBV066, BI655066) isan IgG1 monoclonal antibody targeting the P19 subunit of IL-23, which has been developed for psoriasis. In October 2017, Abbvie announced the completion of phase III trials of ustekinumab and Adalimumab as controls. The effectiveness of Risankizumab was evaluated against ustekinumab and placebo in the ultimma-1 (NCT02684370) and Ultimma-2 (NCT02684357) trials. After 16 weeks of treatment, 75% of risankizumab patients reached PASI90 in both trials, compared with 42% and 48% of Ustekinumab patients and 5% and 2% of placebo patients, respectively. Compliance with sPGA scores was 88% and 84% for risankizumab, 63% and 62% for Ustekinumab, and only 8% and 5% for placebo. In the IMMvent study (NCT02694523), 72% of patients in the Risankizumab group reached PASI90 for 16 weeks, only 47% in the Atamumab group reached PASI90, 84% in the Risankizumab group reached sPGA, and only 60% in the Atamumab group. The product is expected to be submitted to BLA in 2018 and marketed in 2019.
Satralizumab
Satralizumab (SA237) is a human IgG2 monoclonal antibody, targeting the interleukin-6 receptor (IL-6R), which has been developed for neuromyelitis optic or neuromyelitis spectrum disease. NCT02028884, a phase III clinical trial evaluating the safety and efficacy of Satralizumab with baseline treatment, is ongoing. The primary treatment endpoint is the time to first relapse of the disease within 30 months. The trial has enrolled 70 patients and is expected to conclude in July 2018. Another placebo-controlled phase III clinical trial, NCT02073279, has also completed recruitment. The primary treatment end point is the first relapse of the disease in 38 months. The trial has enrolled 98 patients and is expected to end in October 2018. The product has previously been recognized as an orphan drug for NEUROmyelitis spectrum disease by the FDA and EMA, and is expected to be filed for marketing in the second half of 2018.
Brolucizumab
Brolucizumab (RTH258) is A single-chain variable region fragment targeting vascular epidermal cell growth factor A(VEGF) -a, which has been developed for neovascular age-related macular degeneration (nAMD). In June 2017, Novartis announced that HAWK (NCT02307682) and HARRIER (NCT02434328) had reached the end point of treatment, and that Brolucizumab's 48-week average improvement in best-corrected visual acuity was not worse than aflibercept's relative to baseline. In these two trials, more than 1,800 nAMD patients were administered brolucizumab 6mg, Brolucizumab 3mg (not involved in HARRIER) or Aflibercept 2mg, respectively. The results showed that Brolucizumab 6mg reached the primary end point and the critical secondary end point with significant P values in both experiments. Brolucizumab 3mg also reaches these ends in the HAWK. In the HAWK and HARRIER trials, 57 percent and 52 percent of patients who took the drug once 12 weeks followed through to 48 weeks, respectively. Brolucizumab showed good tolerance, with an overall rate of ocular and non-ocular aderse effects comparable to aflibercept. Novartis expects to complete PK trials of low dose specifications in 2018, and BLA is expected to be submitted in the second half of 2018.
PRO140
PRO140 is a humanized IgG4 monoclonal antibody, targeting CC chemokine receptor 5 (CCR5), which can prevent HIV virus from entering T cells. Currently, the developed indication is AIDS. Two phase 2/3 clinical trials (NCT02483078 and NCT02859961) were completed in October 2017 and December 2017. NCT02483078 is a randomized, double-blind, placebo-controlled clinical trial in which drug-resistant HIV patients were treated with PRO140 or placebo in the context of optimal antiviral therapy. NCT02859961, on the other hand, studied the effectiveness of PRO140 alone in patients with stable condition after antiretroviral therapy. In October 2017, CytoDyn met with the FDA to discuss the number and type of patients needed to deliver a BLA combined. The FDA asked the NCT02483078 trial to complete a study of 50 patients and the BLA to submit a safety assessment of a total of 300. The product is currently eligible for FDA rapid review, which means it can submit a rolling BLA application.
Sacituzumab govitecan
Sacituzumab Govitecan (Immu-132) is an antibody conjugate derived from the human anti-trop-2 antibody conjugating the active irinotecan metabolite SN38, and is developed for advanced third-negative breast cancer (TNBC). In November 2017, Immunomedics announced that it would be presenting the BLA in the first quarter of 2018, following the product's FDA breakthrough therapy designation for triple negative breast cancer, supporting the BLA data from the NCT01631552 study. In 110 patients with tri-negative breast cancer who received sacituzumab govitecan at 10mg/kg, the objective response rate was 34%, the clinical benefit rate was 46%, and the kaplan-meier (KM) median expiration and progression-free survival were 7.6 and 5.5 months, respectively. In the phase III clinical trial to verify the efficacy of the product in the treatment of recurrent or refractory tri-negative breast cancer, ASCENT(NCT02574455) recruited 328 patients who progressed after chemotherapy with more than two regimens or at least one chemotherapy regimen and received adjuvant therapy within 12 months. Patients in the trial received sacituzumab Govitecan or chemotherapy with four pre-defined single-drug regimens, which are already under way.
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22, cat-8015) is an antibody conjugate with a molecular weight of 38kDa that fuses the toxic portion of recombinant pseudomonas aeruginosa exotoxin with the variable region fragment of CD22 antibody. Moxetumomab Pasudotox was approved by the FDA as an orphan drug for hairy Cell leukemia (HCL) in February 2016. The product is currently undergoing a multicenter, one-arm phase III clinical trial of relapsed or refractory HCL (NCT01829711), which reached its primary endpoint in May 2017, and Astrazeneca plans to submit the BLA in 2018.
Cemiplimab
Cemiplimab (REGN2810, SAR439684) is a human programmed death receptor 1 (PD-1) monoclonal antibody, developed for inoperable or metastatic skin squamous cell carcinoma (CSCC), which has been recognized as a breakthrough therapy by FDA. Phase I clinical trial NCT02383212 showed that the product had a good effect on both locally advanced and metastatic CSCC, with a comprehensive objective response rate of 46.2%. Currently, Cemiplimab is being jointly developed by Sanofi and Regenerative Yuan, and the BLA is expected to be submitted for the first time in q1 2018. A second phase ii trial, EMPOWER-CSCC1, is also under way in inoperable or metastatic CSCCS. In addition to CSCC, Sanofi and Regenerating unit are actively conducting phase III clinical trials for cervical cancer (NCT03257267) and non-small cell lung cancer (NCT03088540), with the end points reaching May 2020 and November 2021, respectively.
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101) is a glycosylated chimeric antibody targeting CD20. This antibody contains low levels of fucosaccharide, which improves the binding rate of Fc RIIIa, enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) phase bilituximab, developed for chronic lymphoma leukemia (CLL). A multicentre, open-label phase III clinical GENUINE trial (NCT02301156) assessed the safety and efficacy of ublituximab in combination with irutinib versus irutinib alone. A total of 126 CLL patients were enrolled in this trial, who were given irutinib 420mg or irutinib 420mg+ublituximab once a day. The combination group showed an objective remission rate of 78%, while the irutinib alone group showed only 45%. Progression-free survival was also improved, with a risk ratio of 0.559. Another phase III clinical trial, unity-CLL (NCT02612311), was designed to assess the safety and efficacy of Ublituximab in combination with the PI3K inhibitor TGR-1202 for CLL, using obinutuzumab+ benzoate nitrogen and Ublituximab alone or TGR-1202 alone. The trial is already underway and is expected to reach a conclusion in September 2018. In addition to CLL, clinical studies of obinutuzumab combined with TGR-1202 in the treatment of non-Hodgkin's lymphoma are also under way. The trial is a phase 2/3 clinical trial called UnityNHL (NCT02793583) and is expected to reach its endpoint in May 2019. In addition to cancer, Obinutuzumab is also actively developing treatment options for MS. Two randomized, double-blind, multicenter phase III clinical trials ULTIMATEI (NCT03277261) and ULTIMATEII (NCT03277248) have been conducted to compare the safety and efficacy of Obinutuzumab and Terifluramide in relapsed MS. The expected endpoint for both trials is March 2021.
Monoclonal antibodies are in advanced clinical development
In addition to the above mentioned products, 20 monoclonal antibody products are in the post-clinical stage, which are sirukumab, Lampalizumab, Roledumab, emapalumab, fasinumab, tanezumab, etrolizumab, Birtamimab, Gantenerumab, Anifrolumab, tremelimumab, isatuximab, BCD-100, carabab, camrelizumab, IBI308, Glembatumumab Vedotin, Mirvetuximab Soravtansine, oportuzumab Monatox, L19IL2/L19TNF. Sirukumab of them have in September 2017 were refused by the FDA, the FDA asked Johnson added additional clinical data prove sirukumab security, most of the 19 other products haven't key clinical trial data, but these data are expected in the second half of 2018 or 2019, time to market is expected between 2019 and 2020, we'll introduce them surprise until next year.
There is no doubt that the development of monoclonal antibodies is speeding up. It is estimated that the total number of approved monoclonal antibodies will exceed 100 by 2020. Meanwhile, the market of monoclonal antibodies will be booming.
Lanadelumab (SHP643, DX-2930) is a human IgG1 monoclonal antibody that blocks bradykinin production by inhibiting plasma kinidin releasing enzyme. The indication for Lanadelumab in phase III was hereditary angioedema (HAE), a rare condition characterized by tissue edema following exposure. Lanadelumab has been granted orphan eligibility for HAE and FDA breakthrough Therapy for HAE in the U.S. and Europe. In May 2017, Shire reported data from a 26-week phase III clinical HELP trial (NCT02586805) that assessed the safety and efficacy of Lanadelumab in preventing the incidence of acute vascular edema in patients with HAE type I and II. Forty-nine patients were given either 300mg of lanadelumab every two weeks, 300mg of lanadelumab every four weeks, or 150mg of lanadelumab every four weeks or placebo. The results showed that the incidence of angioedema decreased by 87%, 73%, and 76%, respectively, compared with placebo in patients who were given Lanadelumab 300mg once every two weeks, 300mg once every four weeks, and 150mg lanadelumab 150mg once every four weeks, reaching the established primary and secondary endpoints. Another open-label phase III clinical long-term safety and efficacy study (NCT02741596) was conducted to assess the efficacy of Lanadelumab in preventing the development of acute vascular edema in patients with HAE I and II, with the end point expected in February 2018. Marketing applications for this product will be submitted to the FDA and EMA in early 2018.
Crizanlizumab
Crizanlizumab (SEG101) is a humanized monoclonal antibody that targets a platelet-based selective protein known as CD62. The indications studied are sickle cell pain crisis (SCPC). Sickle cell crisis is clinically manifested as chronic hemolytic anemia, prone to infection and recurrent pain crises leading to chronic ischemia and local damage to organs and tissues. In the placebo controlled phase II SUSTAIN test (NCT01895361), 198 patients aged 16 to 65 years were treated with Crizanlizumab every four weeks at a dose of 5mg/kg for 50 consecutive weeks. The median annual rate of SCPC decreased by 45.3%(1.63 vs 2.98), and the time of first OCCURRENCE of SCPC was 2.9 times longer than that of placebo (4.07 vs 1.38 months). The second occurrence of SCPC was 2.0 times longer than placebo (median 10.32 months and 5.09 months, respectively). Previously, Crizanlizumab has been certified as an ORPHAN SCPC in the United States and Europe, and novartis will submit the BLA in 2018 if the PK/PD data is consistent with the final manufacturing process.
Ravulizumab
Ravulizumab (ALXN1210), a humanized monoclonal antibody targeting complement 5(C5), is the next generation of Eculizumab, currently in the pre-registration stage. Its clinical trials included two phase III trials associated with paroxysmal haemoglobinuria (PNH) and two phase III trials associated with atypical hemolytic uremic syndrome (aHUS). These two (PNH) phase III clinical trials NCT02946463 and NCT03056040 are based on the results of a phase 1/2 clinical trial. Plasma lactate dehydrogenase (LDH) levels decreased rapidly and continuously after Ravulizumab in patients without complement inhibitor treatment, and FACIT fatigue scale scores were significantly improved. Phase 3 clinical trial NCT02946463 was designed to assess the safety and efficacy of Ravulizumab versus Eculizumab in adult PNH patients not treated with a complement inhibitor. In this trial, patients were given a loading dose on the first day based on body weight, and maintenance dose on the 15th day (40 to 60kg body weight: loading dose of 2400mg, maintenance dose of 3000mg every 8 weeks; 60 to 100kg body weight: 2700mg loading dose, 3300mg maintenance dose every 8 weeks; For patients with body weight greater than 100kg, the loading dose was 3000mg and the maintenance dose was 3600mg every 8 weeks), while eculizumab was administered according to the instructions. The expected clinical end point was a normal level of plasma lactate dehydrogenase (LDH) within 26 weeks. The trial enrolled 246 patients and will end in December 2017. Another phase 3 trial was designed to evaluate Ravulizumab in stable patients treated with Eculizumab for more than 6 months. Ravulizumab patients were given the same regimen as in the previous trial, while eculizumab patients were given a loading dose of Ravulizumab on Day 183 and a maintenance dose on day 197, with the primary end point being changes in plasma lactate dehydrogenase (LDH) levels at 26 weeks. The trial, which enrolled 197 patients, will end in March 2018. Alex is expected to report the results of the two trials in the second quarter of 2018 and submit the BLA. Ravulizumab has previously been recognized as an orphan drug for PNH in the United States and Europe. The single-arm phase III clinical trial associated with aHUS enrolled 55 patients and is expected to conclude in early 2018. Another phase III clinical trial involving children WITH aHUS, NCT03131219, is also underway and will end in December 2018.
Eptinezumab
Eptinezumab (ALD403) is an IgG1 monoclonal antibody targeting a calcitonin gene-related peptide (CGRP) that was developed for migraine. In June 2017, Alder BioPharma announced that the Phase III Clinical PROMISE1 trial (NCT02559895) was achieving a primary and secondary endpoint. The average number of headache days was 8.6 days/month for patients in the study who were given EPtinezumab 300mg, 100mg, 30mg or placebo, while the low-dose 30mg group was not included in the results. The results showed that a significant decrease in the number of headaches per month was observed between weeks 1 and 12, with a monthly headache of 4.3 days in the 300mg group, 3.9 days in the 100mg group, and 3.2 days in the placebo group. Nearly one-third of patients experienced a 75% or more decrease in the number of headache days per month at week 4-12, with an average of one-fifth of patients having no headache for at least one month within 1-6 months. Another Phase 3 clinical trial in PROMISE2 (NCT02974153) specifically designed for chronic migraine has enrolled 1,050 patients and will be completed in the first half of 2018, with Alder planning to file a marketing application in the second quarter of 2018.
Risankizumab
Risankizumab (ABBV066, BI655066) isan IgG1 monoclonal antibody targeting the P19 subunit of IL-23, which has been developed for psoriasis. In October 2017, Abbvie announced the completion of phase III trials of ustekinumab and Adalimumab as controls. The effectiveness of Risankizumab was evaluated against ustekinumab and placebo in the ultimma-1 (NCT02684370) and Ultimma-2 (NCT02684357) trials. After 16 weeks of treatment, 75% of risankizumab patients reached PASI90 in both trials, compared with 42% and 48% of Ustekinumab patients and 5% and 2% of placebo patients, respectively. Compliance with sPGA scores was 88% and 84% for risankizumab, 63% and 62% for Ustekinumab, and only 8% and 5% for placebo. In the IMMvent study (NCT02694523), 72% of patients in the Risankizumab group reached PASI90 for 16 weeks, only 47% in the Atamumab group reached PASI90, 84% in the Risankizumab group reached sPGA, and only 60% in the Atamumab group. The product is expected to be submitted to BLA in 2018 and marketed in 2019.
Satralizumab
Satralizumab (SA237) is a human IgG2 monoclonal antibody, targeting the interleukin-6 receptor (IL-6R), which has been developed for neuromyelitis optic or neuromyelitis spectrum disease. NCT02028884, a phase III clinical trial evaluating the safety and efficacy of Satralizumab with baseline treatment, is ongoing. The primary treatment endpoint is the time to first relapse of the disease within 30 months. The trial has enrolled 70 patients and is expected to conclude in July 2018. Another placebo-controlled phase III clinical trial, NCT02073279, has also completed recruitment. The primary treatment end point is the first relapse of the disease in 38 months. The trial has enrolled 98 patients and is expected to end in October 2018. The product has previously been recognized as an orphan drug for NEUROmyelitis spectrum disease by the FDA and EMA, and is expected to be filed for marketing in the second half of 2018.
Brolucizumab
Brolucizumab (RTH258) is A single-chain variable region fragment targeting vascular epidermal cell growth factor A(VEGF) -a, which has been developed for neovascular age-related macular degeneration (nAMD). In June 2017, Novartis announced that HAWK (NCT02307682) and HARRIER (NCT02434328) had reached the end point of treatment, and that Brolucizumab's 48-week average improvement in best-corrected visual acuity was not worse than aflibercept's relative to baseline. In these two trials, more than 1,800 nAMD patients were administered brolucizumab 6mg, Brolucizumab 3mg (not involved in HARRIER) or Aflibercept 2mg, respectively. The results showed that Brolucizumab 6mg reached the primary end point and the critical secondary end point with significant P values in both experiments. Brolucizumab 3mg also reaches these ends in the HAWK. In the HAWK and HARRIER trials, 57 percent and 52 percent of patients who took the drug once 12 weeks followed through to 48 weeks, respectively. Brolucizumab showed good tolerance, with an overall rate of ocular and non-ocular aderse effects comparable to aflibercept. Novartis expects to complete PK trials of low dose specifications in 2018, and BLA is expected to be submitted in the second half of 2018.
PRO140
PRO140 is a humanized IgG4 monoclonal antibody, targeting CC chemokine receptor 5 (CCR5), which can prevent HIV virus from entering T cells. Currently, the developed indication is AIDS. Two phase 2/3 clinical trials (NCT02483078 and NCT02859961) were completed in October 2017 and December 2017. NCT02483078 is a randomized, double-blind, placebo-controlled clinical trial in which drug-resistant HIV patients were treated with PRO140 or placebo in the context of optimal antiviral therapy. NCT02859961, on the other hand, studied the effectiveness of PRO140 alone in patients with stable condition after antiretroviral therapy. In October 2017, CytoDyn met with the FDA to discuss the number and type of patients needed to deliver a BLA combined. The FDA asked the NCT02483078 trial to complete a study of 50 patients and the BLA to submit a safety assessment of a total of 300. The product is currently eligible for FDA rapid review, which means it can submit a rolling BLA application.
Sacituzumab govitecan
Sacituzumab Govitecan (Immu-132) is an antibody conjugate derived from the human anti-trop-2 antibody conjugating the active irinotecan metabolite SN38, and is developed for advanced third-negative breast cancer (TNBC). In November 2017, Immunomedics announced that it would be presenting the BLA in the first quarter of 2018, following the product's FDA breakthrough therapy designation for triple negative breast cancer, supporting the BLA data from the NCT01631552 study. In 110 patients with tri-negative breast cancer who received sacituzumab govitecan at 10mg/kg, the objective response rate was 34%, the clinical benefit rate was 46%, and the kaplan-meier (KM) median expiration and progression-free survival were 7.6 and 5.5 months, respectively. In the phase III clinical trial to verify the efficacy of the product in the treatment of recurrent or refractory tri-negative breast cancer, ASCENT(NCT02574455) recruited 328 patients who progressed after chemotherapy with more than two regimens or at least one chemotherapy regimen and received adjuvant therapy within 12 months. Patients in the trial received sacituzumab Govitecan or chemotherapy with four pre-defined single-drug regimens, which are already under way.
Moxetumomab pasudotox
Moxetumomab pasudotox (HA22, cat-8015) is an antibody conjugate with a molecular weight of 38kDa that fuses the toxic portion of recombinant pseudomonas aeruginosa exotoxin with the variable region fragment of CD22 antibody. Moxetumomab Pasudotox was approved by the FDA as an orphan drug for hairy Cell leukemia (HCL) in February 2016. The product is currently undergoing a multicenter, one-arm phase III clinical trial of relapsed or refractory HCL (NCT01829711), which reached its primary endpoint in May 2017, and Astrazeneca plans to submit the BLA in 2018.
Cemiplimab
Cemiplimab (REGN2810, SAR439684) is a human programmed death receptor 1 (PD-1) monoclonal antibody, developed for inoperable or metastatic skin squamous cell carcinoma (CSCC), which has been recognized as a breakthrough therapy by FDA. Phase I clinical trial NCT02383212 showed that the product had a good effect on both locally advanced and metastatic CSCC, with a comprehensive objective response rate of 46.2%. Currently, Cemiplimab is being jointly developed by Sanofi and Regenerative Yuan, and the BLA is expected to be submitted for the first time in q1 2018. A second phase ii trial, EMPOWER-CSCC1, is also under way in inoperable or metastatic CSCCS. In addition to CSCC, Sanofi and Regenerating unit are actively conducting phase III clinical trials for cervical cancer (NCT03257267) and non-small cell lung cancer (NCT03088540), with the end points reaching May 2020 and November 2021, respectively.
Ublituximab
Ublituximab (LFB-R603, TGT-1101, TGTX-1101) is a glycosylated chimeric antibody targeting CD20. This antibody contains low levels of fucosaccharide, which improves the binding rate of Fc RIIIa, enhanced antibody-dependent cell-mediated cytotoxicity (ADCC) phase bilituximab, developed for chronic lymphoma leukemia (CLL). A multicentre, open-label phase III clinical GENUINE trial (NCT02301156) assessed the safety and efficacy of ublituximab in combination with irutinib versus irutinib alone. A total of 126 CLL patients were enrolled in this trial, who were given irutinib 420mg or irutinib 420mg+ublituximab once a day. The combination group showed an objective remission rate of 78%, while the irutinib alone group showed only 45%. Progression-free survival was also improved, with a risk ratio of 0.559. Another phase III clinical trial, unity-CLL (NCT02612311), was designed to assess the safety and efficacy of Ublituximab in combination with the PI3K inhibitor TGR-1202 for CLL, using obinutuzumab+ benzoate nitrogen and Ublituximab alone or TGR-1202 alone. The trial is already underway and is expected to reach a conclusion in September 2018. In addition to CLL, clinical studies of obinutuzumab combined with TGR-1202 in the treatment of non-Hodgkin's lymphoma are also under way. The trial is a phase 2/3 clinical trial called UnityNHL (NCT02793583) and is expected to reach its endpoint in May 2019. In addition to cancer, Obinutuzumab is also actively developing treatment options for MS. Two randomized, double-blind, multicenter phase III clinical trials ULTIMATEI (NCT03277261) and ULTIMATEII (NCT03277248) have been conducted to compare the safety and efficacy of Obinutuzumab and Terifluramide in relapsed MS. The expected endpoint for both trials is March 2021.
Monoclonal antibodies are in advanced clinical development
In addition to the above mentioned products, 20 monoclonal antibody products are in the post-clinical stage, which are sirukumab, Lampalizumab, Roledumab, emapalumab, fasinumab, tanezumab, etrolizumab, Birtamimab, Gantenerumab, Anifrolumab, tremelimumab, isatuximab, BCD-100, carabab, camrelizumab, IBI308, Glembatumumab Vedotin, Mirvetuximab Soravtansine, oportuzumab Monatox, L19IL2/L19TNF. Sirukumab of them have in September 2017 were refused by the FDA, the FDA asked Johnson added additional clinical data prove sirukumab security, most of the 19 other products haven't key clinical trial data, but these data are expected in the second half of 2018 or 2019, time to market is expected between 2019 and 2020, we'll introduce them surprise until next year.
There is no doubt that the development of monoclonal antibodies is speeding up. It is estimated that the total number of approved monoclonal antibodies will exceed 100 by 2020. Meanwhile, the market of monoclonal antibodies will be booming.
Monoclonal antibodies are at the end of the clinical stage

Chromatin A lot of references were given in the paper TO Watch in 2018. However, this paper was not a translated paper. Chromatin was carried out on the basis of the chromatin, the information was further added or truncated, and chromatin IP addresses and the author's opinions were added. In addition, this article is only published for study and exchange, reprint this article must indicate the author and source, otherwise it will be regarded as malicious infringement, infringement shall be corrected. For those who disagree, please refer to the Original English text. Address:
reference:Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/

reference:Hélène Kaplon, Janice M. Reichert. Antibodies to watch in 2018[EB/OL].(2018-01-04)[2018-01-09] :http://www.antibodysociety.org/author/janreichert/
address:NO.106 Fenghe Road Beibei District,Chongqing
pegbio@pegbiocq.com
Search
NameDescriptionContent
PEG-BIO Biopharm Co,.(Chongqing) All right reserved 渝ICP备18012241号

contact us
15823078768 Mr.LIU